The effect number of SNPs.
使用simpleM计算有效标记数
- 基因型数据插补
- 将字符型基因型转换为数字格式
- 使用simpleM计算有效标记数
simpleM(http://simplem.sourceforge.net/)是一个使用R编写的、使用相关SNP进行遗传关联研究的多重测试校正方法。对于低密度的SNP标记的基因型可以计算有效标记数。结合相关方法(0.05/N, N为有效标记数)对GWAS的结果进行P值矫正。因其对数据输入格式有简单的要求,我们可以按照如下方法进行处理。
1 使用Tassel插补缺失基因型
- 打开 Tassel (Version:5.2.78)5.0 以上都可以。我这里用的最新版。

- 导入基因型后使用LD KNNi 方法进行缺失基因型插补,使用默认参数即可
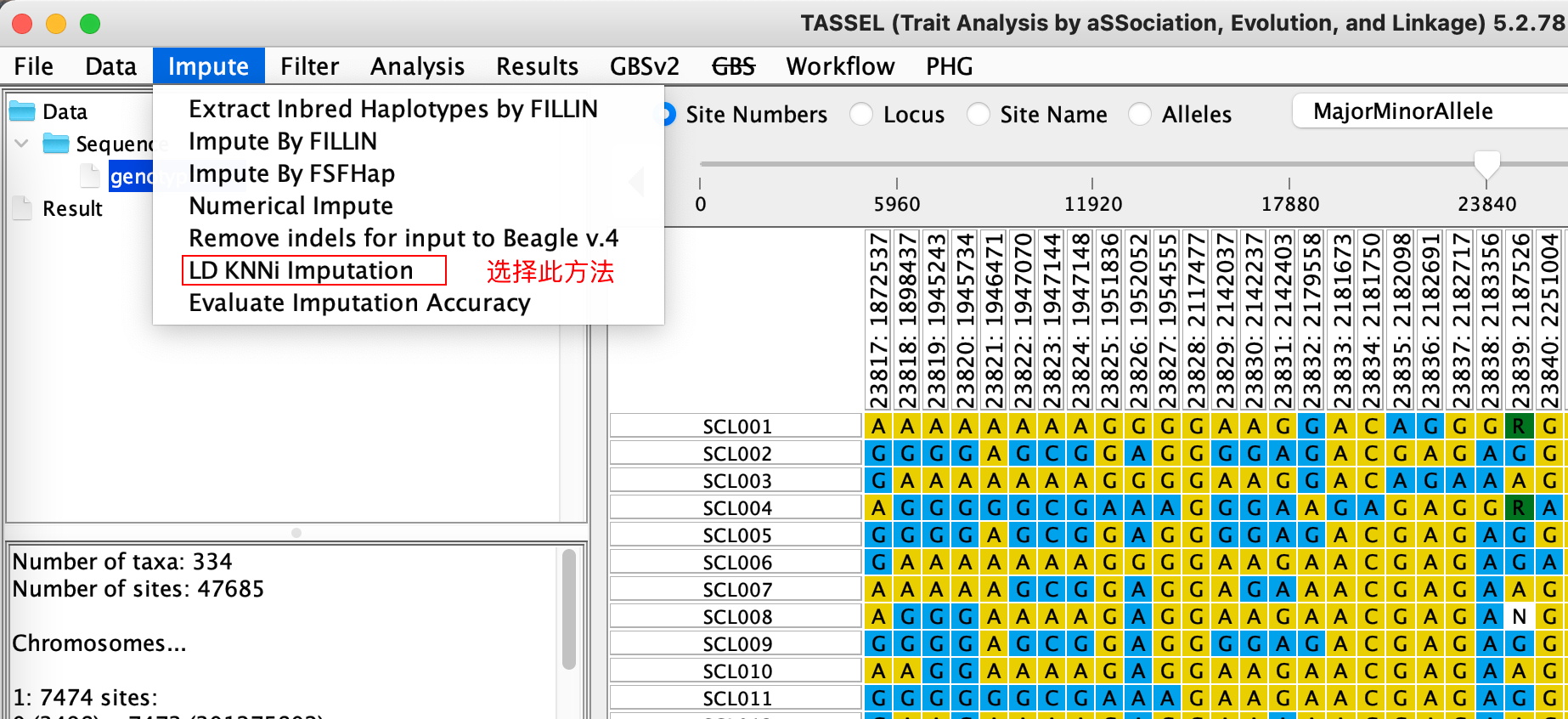
插补方法有多种,由于simpleM需要无缺失的基因型数据,所以大家可以自行选择插补方法进行缺失基因型的插补。
2 基因型转换为数字格式
1). 使用Tassel将字符型基因型格式转换为数字格式 操作步骤如下。需要的主要的是Tassel转化出来的基因型中只有0,0.5,1三种数字,simpleM要求是0,1,2,所以还需要在R中进行适当转化,也可以在excel进行处理。
Genotypes are coded as 0, 1 and 2: 0, 1 and 2 are the number of the reference alleles. Genotypes are separated by one-character white spaces. Rows are SNPs and columns are individuals. SNPs should be in their physical order (for calculating LD). NO SNP names and individuals IDs. Missing values should be imputed, which is mainly for keeping the correlation matrix positive semi-definite. There are several possible ways to fill in the missing values, e.g. using imputation software, K-nearest neighbor (KNN) or replacing them with the common allele genotypes.
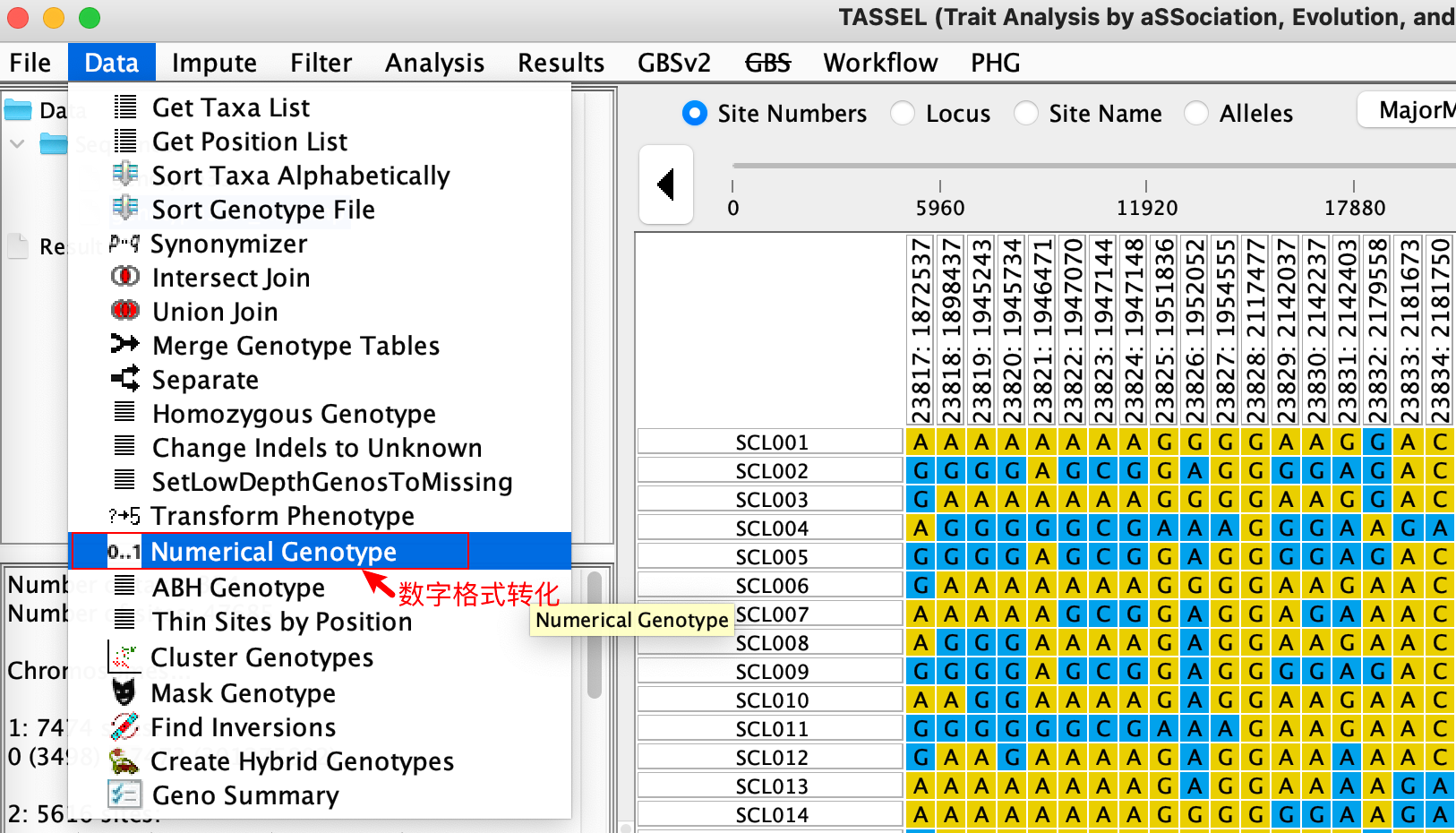
Tassel转换出的数字中1,0.5,0分别表示主要等位位点,杂合位点,及次要等位位点。

2). 导出转换后的基因型 选中转换后的基因型,然后在选择 File–>Save as, 选择Numeric Genotype格式进行输出。
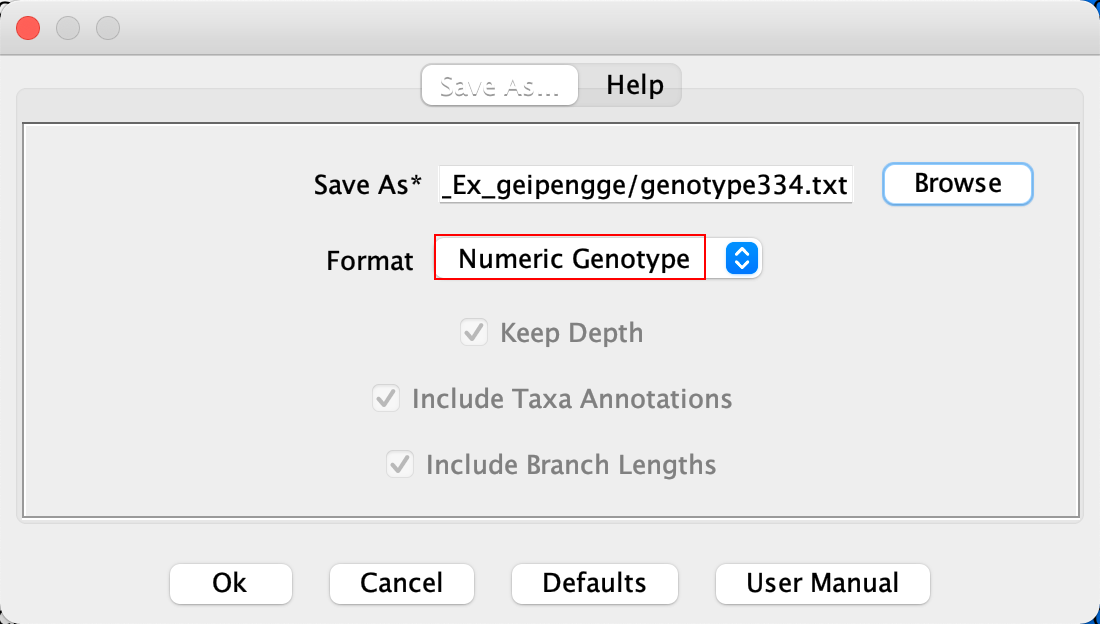
这里也可以R语言GAPIT软件包对基因型进行转化。
3 使用simpleM计算有效标记数
Tassel输出的数字格式基因型带有
# 以下部分为 simpleM 源代码
Meff_PCA <- function(eigenValues, percentCut){
totalEigenValues <- sum(eigenValues)
myCut <- percentCut*totalEigenValues
num_Eigens <- length(eigenValues)
myEigenSum <- 0
index_Eigen <- 0
for(i in 1:num_Eigens){
if(myEigenSum <= myCut){
myEigenSum <- myEigenSum + eigenValues[i]
index_Eigen <- i
}
else{
break
}
}
return(index_Eigen)
}
#============================================================================
# infer the cutoff => Meff
inferCutoff <- function(dt_My){
CLD <- cor(dt_My)
eigen_My <- eigen(CLD)
# PCA approach
eigenValues_dt <- abs(eigen_My$values)
Meff_PCA_gao <- Meff_PCA(eigenValues_dt, PCA_cutoff)
return(Meff_PCA_gao)
}
#=============================================================
PCA_cutoff <- 0.995
#=============================================================
# 以下代码需要根据自己的文件进行修改
# setwd("/Users/alipe/Documents/05.Project/06.GWAS_QTL/GWAS/simpleM")
# Genotype_KNNlmp.txt 为刚刚Tassel输出的数字格式的基因型(已删除<Numberic>表头)
tassel_numer_format <- read.delim("Genotype_KNNlmp.txt", header = T, row.names = 1)
simplem_in <- as.matrix(t(tassel_numer_format * 2))
# 如果输出的数字基因型中还存在NA等字符,需要删除,或者替换
simplem_in <- na.omit(simplem_in)
dim(simplem_in)
write.table(x = simplem_in, file = "simpleM_in.txt", quote = F,
row.names = F, col.names = F, sep = "\t")
## ======================================================= ##
# 以下代码开始计算有效标记数
fn_In <- "simpleM_in.txt" # <---- change path here!!!
mySNP_nonmissing <- read.table(fn_In, colClasses="integer")
numLoci <- length(mySNP_nonmissing[, 1])
simpleMeff <- NULL
fixLength <- 133
i <- 1
myStart <- 1
myStop <- 1
while(myStop < numLoci){
myDiff <- numLoci - myStop
if(myDiff <= fixLength) break
myStop <- myStart + i*fixLength - 1
snpInBlk <- t(mySNP_nonmissing[myStart:myStop, ])
MeffBlk <- inferCutoff(snpInBlk)
simpleMeff <- c(simpleMeff, MeffBlk)
myStart <- myStop+1
}
snpInBlk <- t(mySNP_nonmissing[myStart:numLoci, ])
MeffBlk <- inferCutoff(snpInBlk)
simpleMeff <- c(simpleMeff, MeffBlk)
# 查看结果
cat("Total number of SNPs is: ", numLoci, "\n")
cat("Inferred Meff is: ", sum(simpleMeff), "\n")
# 输出矫正阈值
print(0.05/sum(simpleMeff))
一般可能会有如下报错:
Error in eigen(CLD) : infinite or missing values in 'x'
In addition: Warning message:
In cor(dt_My) : the standard deviation is zero
Called from: eigen(CLD)
原因可能是由于数据存在非多态性SNP位点导致的。建议在Tassel导入基因型后就进行过滤。
结果如下:
> cat("Total number of SNPs is: ", numLoci, "\n")
Total number of SNPs is: 38732
> cat("Inferred Meff is: ", sum(simpleMeff), "\n")
Inferred Meff is: 23085
> print(0.05/sum(simpleMeff))
[1] 2.165909e-06
参考链接:
- simpleM说明:http://simplem.sourceforge.net/
- SNP多重检验矫正方法:https://doi.org/10.1002/gepi.20563